[ad_1]
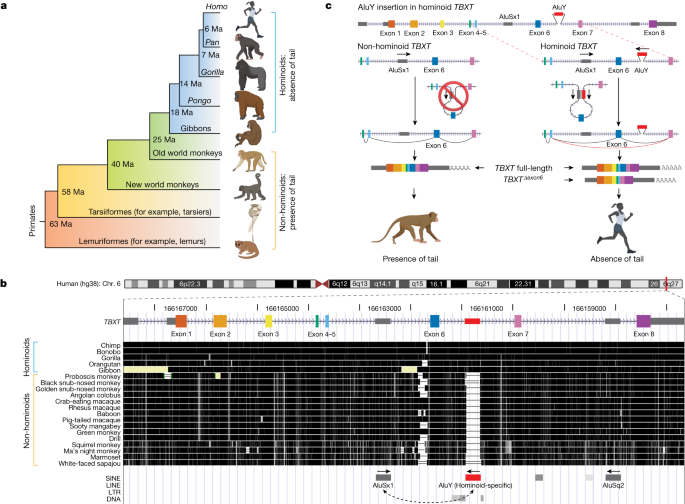
Comparative genomics analyses of tail-development-related genes
Hominoid evolution represents an prolonged stage in primate evolution that concerned many phenotypic modifications and widespread genomic sequence modifications. Due to this fact, querying for hominoid-specific mutations throughout the genome leads to tens of hundreds of thousands of candidates, with most of them disposed in non-coding areas. We used the next standards to outline {that a} candidate variant might contribute to the tail-loss evolution in hominoids: (1) needs to be hominoid-specific, which implies that the variant sequence or amino acid is exclusive to hominoid species and can’t be shared by some other species which have tails; (2) the operate of the related genes pertains to tail improvement. Tail-development-related genes in vertebrates have been collected from the MGI phenotype database and extra literature not lined by the MGI database. The preliminary analyses primarily lined genes extracted from the MGI time period MP0003456 for ‘absent tail’ phenotype (https://www.informatics.jax.org/vocab/mp_ontology/MP:0003456), to a complete of 31 genes. Extra analyses included genes from MP0002632 for ‘vestigial tail’ (https://www.informatics.jax.org/vocab/mp_ontology/MP:0002632) and MP0003456 for ‘quick tail’ (https://www.informatics.jax.org/vocab/mp_ontology/MP:0000592). Collectively, the ultimate listing of genes associated to vertebrate tail improvement included 140 genes (as of MGI updates in February 2023) and the mutations of that are reported to be associated to tail-reduction phenotypes (Supplementary Information 1).
Gene construction annotations of the 140 genes have been downloaded from BioMart of Ensembl 109 (https://useast.ensembl.org/info/data/biomart/index.html). The longest transcript with probably the most exons have been chosen for every gene. Multiz30way alignments of genomic sequences throughout 27 primate species have been downloaded from the UCSC Genome Browser. We chosen all six hominoid species (hg38, gorGor5, panTro5, panPan2, ponAbe2 and nomLeu3) to calculate a hominoid-consensus sequence, and used two non-hominoid species (pig-tailed macaque, macNem1, and marmoset, calJac3) because the outgroups. The homologous areas of the 140 genes, along with 10,000 bp each upstream and downstream sequences, within the 8 species have been extracted from Multiz30way alignment utilizing bedtools (v.2.30.0)52. Hominoid-specific variants have been recognized utilizing the next parameters: SNVs or substitutions shared by six hominoid species however completely different in any of the 2 outgroup monkey species have been recognized as putative hominoid-specific SNVs (Supplementary Information 2); DNA sequences current in all six hominoid species however absent in both of the 2 outgroup monkey species have been recognized as hominoid-specific insertions (Supplementary Information 3); and DNA sequences absent from all six hominoid species however current in each of the 2 outgroup monkey species have been recognized as hominoid-specific deletions (Supplementary Information 4). Notably, our standards for analysing hominoid-specific variants might embrace a small proportion of false-positive hits which might be outgroup-specific variants.
We used the Ensembl variant impact predictor (built-in in Ensembl 109)53 to deduce the potential useful impression of the detected hominoid-specific SNVs, insertions and deletions. Owing to the dearth of an ancestral genome because the reference sequence, variant impact predictor predictions have been carried out inversely utilizing the human/hominoid genomic sequence because the reference allele, and the outgroup sequence served as the choice allele. SNVs annotated as both ‘deleterious’ (<0.05) within the SIFT rating or ‘damaging’ (>0.446) within the PolyPhen rating (53 cases), and insertions (6 cases) or deletions (2 cases) that have an effect on protein sequences have been collected for additional handbook inspection comparability throughout species. This extra inspection was carried out throughout the Cactus Alignment of the genomes throughout 241 species within the UCSC Genome Browser Comparative Genomics module51. This inspection discovered that many of the annotated variants that will have an effect on host gene operate fell into three classes: (1) outgroup-specific variants; (2) false-positive annotation of the variant operate in a minor transcript; and (3) missense variants in hominoid species however sharing the identical amino acid in at the very least one different tailed species. These variants weren’t thought of as candidates that will have affected tail-loss evolution in hominoids. Excluding these variants, we recognized 9 variants as true hominoid-specific coding area mutations, together with seven SNVs and two insertions and deletions (Supplementary Information 1). Following identification of prime candidates, protein sequence alignments throughout consultant vertebrate species have been downloaded from the NCBI database and analysed utilizing the MUSCLE algorithm with MEGA X software program and default settings54.
RNA secondary construction prediction
RNA secondary construction prediction of the human TBXT exon 5–intron 5–exon 6–intron 6–exon 7 sequence was carried out utilizing RNAfold (v.2.6.0) via the ViennaRNA Net Server (http://rna.tbi.univie.ac.at/)35. The algorithm calculates the folding likelihood utilizing a minimal free vitality matrix with default parameters. As well as, the calculation included the partition operate and base pairing likelihood matrix. Notably, human TBXT transcripts have been annotated to have a 5′ untranslated area exon, making its exon numbers differ from most of different species, together with mouse. To simplify, we referred to the primary coding exon of human TBXT as exon 1 and thus the choice spliced exon as exon 6, in keeping with mouse Tbxt. RNA secondary construction prediction used the DNA sequence from exon 5 to exon 7 following this order.
Human ES cell tradition and differentiation
Human ES cells (WA01, additionally referred to as H1, from WiCell Analysis Institute) have been authenticated by the distributor WiCell utilizing quick tandem repeat profiling to authenticate the cell traces. Human ES cells have been cultured in feeder-free circumstances on tissue-culture-grade plates coated with human ES cell-qualified Geltrex (Gibco, A1413302). Geltrex was 1:100 diluted in DMEM/F-12 (Gibco, 11320033) supplemented with 1× Glutamax (100X, Gibco, 35050061) and 1% penicillin–streptomycin (Gibco, 15070063). Earlier than seeding human ES cells, the plate was handled with Geltrex working answer in a tissue tradition incubator (37 °C and 5% CO2) for at the very least 1 h.
StemFlex medium (Gibco, A3349401) was used for human ES cell upkeep and culturing in a feeder-free situation based on the producer’s protocol. Briefly, StemFlex full medium was made by combining StemFlex basal medium (450 ml) with 50 ml of StemFlex complement (10×) plus 1% penicillin–streptomycin. Every Geltrex-coated nicely on a 6-well plate was seeded with 200,000 cells to acquire about 80% confluence in 3–4 days. Human ES cells have been cryopreserved in PSC Cryomedium (Gibco, A2644601). The tradition medium was supplemented with 1× RevitaCell (100×, Gibco, A2644501, which can be included within the PSC Cryomedium equipment) when cells had gone via pressured circumstances, akin to freezing-and-thawing or nucleofection. RevitaCell supplemented medium was changed with common StemFlex full medium on the second day. Human ES cells grown beneath the RevitaCell situation may grow to be stretched however would get well after returning to the conventional StemFlex full medium. All human ES cell traces examined detrimental throughout our routine quantitative PCR-based mycoplasma assessments.
The human ES cell differentiation assay to induce a gene expression sample of the primitive streak state was tailored from a beforehand printed methodology36. On day −1, freshly cultured human ES cell colonies have been dissociated into clumps (2–5 cells) utilizing Versene buffer (with EDTA, Gibco, 15040066). The dissociated cells have been seeded on Geltrex-coated 6-well tissue tradition plates at 25,000 cells per cm2 (0.25 M per nicely within the 6-well plates) in StemFlex full medium. Differentiation to the primitive streak state was initiated on the following day (day 0) by switching StemFlex full medium to basal differentiation medium. Basal differentiation medium (50 ml) was made utilizing 48.5 ml DMEM/F-12, 1% Glutamax (500 µl), 1% ITS-G (500 µl, Gibco, 41400045) and 1% penicillin–streptomycin (500 µl), and supplemented with 3 µM GSK3 inhibitor CHIR99021 (10 µl of 15 mM inventory answer in DMSO; Tocris, 4423). The cells have been collected at differentiation day 1 to three for downstream experiments, which confirmed the expression fluctuations of mesoderm genes (TBXT and MIXL1) in a 3-day differentiation interval36 (Prolonged Information Fig. 3).
Mouse ES cell tradition and differentiation
The mouse ES cell line (MK6) derived from the C57BL/6J mouse pressure was obtained from the NYU Langone Well being Rodent Genetic Engineering Laboratory. The wild-type MK6 mouse ES cell line was authenticated by its competence for contributing to embryos when cultured on feeder-cell-dependent circumstances adopted by blastomere injection. MK6 mouse ES cells used on this examine have been cultured in each feeder-dependent and feeder-free tradition circumstances relying on the needs of the experiment. All mouse ES cell traces examined detrimental throughout our routine quantitative PCR-based mycoplasma assessments. For feeder-dependent mouse ES cell tradition circumstances, mouse ES cells have been plated on a pre-seeded monolayer of mouse embryonic fibroblast (MEF) cells (CellBiolabs, CBA-310). MEF-coated plates have been ready by seeding 50,000 cells per cm2 in tissue tradition plates handled with 0.1% gelatin answer (EMD Millipore, ES-006-B). MEF culturing medium was comprised of DMEM (Gibco, 11965118) with 10% FBS (GeminiBio, 100–500), 0.1 mM MEM non-essential amino acids (Gibco, 11140050), 1× Glutamax (Gibco, 35050061) and 1% penicillin–streptomycin (Gibco, 15070063). Mouse ES cell medium was comprised of Knockout DMEM (Gibco, 10829018) containing 15% (v/v) FBS (Hyclone, SH30070.03), 0.1 mM β-mercaptoethanol (Gibco, 31350010), 1× MEM non-essential amino acids (Gibco, 11140050), 1× Glutamax (Gibco, 35050061), 1× nucleosides (Millipore, ES-008-D) and 1,000 models ml–1 LIF (EMD Millipore, ESG1107).
For feeder-free mouse ES cell tradition circumstances, cells have been grown on tissue-culture-grade plates that have been pre-coated with mouse ES cell-qualified 0.1% gelatin (EMD Millipore, ES-006-B) at room temperature for at the very least 30 min. Earlier than seeding mouse ES cells, feeder-free mouse ES cell culturing medium was added to a gelatin-treated plate and warmed in a 37 °C and 5% CO2 incubator for at the very least 30 min. Feeder-free mouse ES cell culturing medium, additionally referred to as ‘80/20’ medium, includes 80% 2i medium and 20% of the above-mentioned mouse ES cell medium by quantity. The 2i medium was comprised of a 1:1 mixture of Superior DMEM/F-12 (Gibco, 12634010) and Neurobasal-A (Gibco, 10888022), adopted by including 1× N2 complement (Gibco, 17502048), 1× B-27 complement (Gibco, 17504044), 1× Glutamax (Gibco, 35050061), 0.1 mM β-mercaptoethanol (Gibco, 31350010), 1,000 models ml–1 LIF (Millipore, ESG1107), 1 µM MEK1/2 inhibitor (Stemgent, PD0325901) and three µM GSK3 inhibitor CHIR99021 (Tocris, 4423).
The mouse ES cell differentiation protocol for inducing Tbxt gene expression was tailored from a beforehand described methodology in a feeder cell-free situation37. Cells have been first plated in 80/20 medium for twenty-four h on a gelatin-coated 6-well plate, adopted by switching to N2/B27 medium with out LIF or 2i for an additional 2 days of tradition. The N2/B27 medium (50 ml) was made with 18 ml Superior DMEM/F-12, 18 ml Neurobasal-A, 9 ml Knockout DMEM, 2.5 ml Knockout Serum Substitute (Gibco, 10828028), 0.5 ml N2 complement, 1 ml B27 complement, 0.5 ml Glutamax (100×), 0.5 ml nucleosides (100×) and 0.1 mM β-mercaptoethanol. Then the N2/B27 medium was supplemented with 3 µM GSK3 inhibitor CHIR99021 to induce differentiation (day 0). The cells have been collected at differentiation day–3 for downstream experiments, which confirmed constant outcomes of Tbxt gene expression fluctuations in a 3-day differentiation interval.
CRISPR concentrating on
All information RNAs of the CRISPR experiments have been designed utilizing the CRISPOR algorithm built-in within the UCSC Genome Browser55. Information RNAs have been cloned into the pX459V2.0-HypaCas9 plasmid (AddGene, plasmid 62988) or its customized by-product by changing the puromycin-resistance gene with the blasticidin-resistance gene. Information RNAs on this examine have been designed in pairs to delete the intervening sequences. Insertion websites for the AluSx1 and AluY pair in mouse Tbxt (TbxtinsASAY) have been chosen by the information RNA high quality and the relative distance in comparison with the human TBXT gene construction. The insertion website for the RCS aspect (TbxtinsRCS) was the identical as for insertion of the AluY aspect. The CRISPR-targeting websites and information RNA sequences are listed in Supplementary Desk 2.
All oligonucleotides (plus Golden-Gate meeting overhangs) have been synthesized by Built-in DNA Applied sciences (IDT) and ligated into an empty pX459V2.0 vector following the usual Golden Gate Meeting protocol utilizing BbsI restriction enzyme (NEB, R3539). The constructed plasmids have been purified from 3 ml Escherichia coli cultures utilizing a ZR Plasmid MiniPrep Purification equipment (Zymo Analysis, D4015) for sequence verification. Plasmids for delivering into ES cells have been purified from 250 ml E. coli cultures utilizing a PureLink HiPure Plasmid Midiprep equipment (Invitrogen, K210005). To facilitate DNA supply to ES cells via nucleofection, the purified plasmids have been resolved in Tris-EDTA buffer (pH 7.5) to a focus of at the very least 1 µg µl–1 in a sterile hood.
DNA supply
DNA supply into human or mouse ES cells for CRISPR–Cas9 concentrating on was carried out utilizing a Nucleofector 2b machine (Lonza, BioAAB-1001). A Human Stem Cell Nucleofector equipment 1 (VPH-5012) and a mouse ES cell Nucleofector equipment (Lonza, VVPH-1001) have been used for delivering DNA into human and mouse ES cells, respectively. ES cells have been double-fed the day earlier than the nucleofection experiment to keep up a superior situation.
Earlier than performing nucleofection on human ES cells, 6-cm tissue tradition plates have been handled with 0.5 µg cm–2 rLaminin-521 in a 37 °C and 5% CO2 incubator for at the very least 2 h. rLaminin-521-treated plates present the perfect viability when seeding human ES cells as single cells. Cultured human ES cells have been washed with DPBS and dissociated into single cells utilizing TrypLE Choose Enzyme (no phenol pink; Gibco, 12563011). A million human ES cell single cells have been nucleofected utilizing program A-023 based on the producer’s directions for the Nucleofector 2b machine. Transfected cells have been transferred onto the rLaminin-521-treated 6-cm plates with pre-warmed StemFlex full medium supplemented with 1× RevitaCell however not penicillin–streptomycin. Antibiotic choice was carried out 24 h after nucleofection with puromycin (ultimate focus of 0.8 μg ml–1; Gibco, A1113802).
Mouse ES cells have been dissociated into single cells utilizing StemPro Accutase (Gibco, A1110501), and 5 million cells have been transfected utilizing program A-023 based on the producer’s directions. Exon 6 deletion in mouse ES cells was carried out utilizing cells cultured within the feeder-free situation. Nucleofected cells have been plated on 0.1% gelatin-treated 10-cm plates, adopted by antibiotic choice 24 h after nucleofection with blasticidin (ultimate focus of seven.5 µg ml–1; Gibco, A1113903). The insertion of the AluSx1–AluY pair and insRCS engineering have been carried out utilizing mouse ES cells cultured on a feeder-dependent situation. Mouse ES cells have been plated on a monolayer of MEF cells seeded on 0.1% gelatin-treated 10-cm plates, adopted by antibiotic choice 24 h after nucleofection.
Along with the pX459V2.0-HypaCas9-gRNA plasmids for nucleofection, single-strand DNA oligonucleotides have been co-delivered for microhomology-induced deletion of the focused sequences56. These ssDNA sequences have been synthesized by IDT via its Ultramer DNA Oligo service, together with three phosphorothioate bond modifications on every finish. Detailed sequence info of those lengthy ssDNA oligonucleotides are listed in Supplementary Desk 3.
For TbxtinsASAY and TbxtinsRCS engineering, homology-based repairing template plasmids, together with a range marker gene puro–ΔTK, (puromycin-resistance gene for optimistic choice and ΔTK, a truncated model of herpes simplex virus sort 1 thymidine kinase, for detrimental choice, as introduced in Prolonged Information Fig. 7), was transfected along with the pX459V2.0-HypaCas9-gRNA plasmids. Following nucleofection and antibiotic choice (0.8 μg ml–1 puromycin for 3 days ranging from the second day of nucleofection), single clones have been picked, adopted by PCR genotyping of CRISPR–Cas9-targeted loci (exon 6 deletion, inserting AluY, inserting AluSx1 or inserting RCS). The genotyping PCR primers are listed in Supplementary Desk 4.
PCR genotyping-confirmed clones have been additional validated utilizing Seize-seq (see beneath) to verify the genotype and to exclude the opportunity of any random integration of plasmid DNA. Subsequently, Cre recombinase was transiently launched to take away the choice marker puro–ΔTK. Cells have been handled with 250 nM ganciclovir for counter-selecting ΔTK-negative cells as the choice marker gene-depleted cells. Following isolation of single mouse ES cell clones of TbxtinsASAY and TbxtinsRCS mouse ES cells with out the choice marker gene, these clones have been used for downstream experiments, together with in vitro differentiation assays and blastocyst injection for producing mouse fashions.
Seize-seq genotyping
Seize-seq, or focused sequencing of the loci of curiosity, was carried out as beforehand described39. Conceptually, Seize-seq makes use of customized biotinylated probes to pull-down the sequences at genomic loci of curiosity from the usual whole-genome sequencing libraries, thereby enabling sequencing of the particular genomic loci in a a lot increased depth whereas decreasing the price.
Genomic DNA was purified from mouse ES cells or from ear punches of mice of curiosity utilizing a Zymo Fast-DNA Miniprep Plus equipment (D4068) based on the producer’s protocol. DNA sequencing libraries appropriate for Illumina sequencers have been ready following the usual protocol. Briefly, 1 µg of gDNA was sheared to 500–900 base pairs in a 96-well microplate utilizing a Covaris LE220 (450 W, 10% obligation issue, 200 cycles per burst and 90-s therapy time), adopted by purification with a DNA Clear and Focus-5 equipment (Zymo Analysis, D4013). Sheared and purified DNA have been then handled with finish restore enzyme combine (T4 DNA polymerase, Klenow DNA polymerase and T4 polynucleotide kinase, NEB, M0203, M0210 and M0201, respectively), and A-tailed utilizing Klenow 3′−5′ exo-enzyme (NEB, M0212). Illumina sequencing library adapters have been subsequently ligated to DNA ends adopted by PCR amplification with KAPA 2X Hello-Fi Hotstart Readymix (Roche, KR0370).
Customized biotinylated probes have been ready as bait via nick translation utilizing BAC DNA and/or plasmids because the template. The probes have been ready to comprehensively cowl the whole locus. We used BAC traces RP24-88H3 and RP23-159G7, bought from BACPAC Genomics, to generate bait probes overlaying the mouse Tbxt locus and about 200 kb flanking sequences in each upstream and downstream areas. The pooled whole-genome sequencing libraries have been hybridized with the biotinylated baits in answer and purified utilizing streptavidin-coated magnetic beads. Following pull-down, DNA sequencing libraries have been quantified utilizing a Qubit 3.0 Fluorometer (Invitrogen, Q33216) with a dsDNA HS Assay equipment (Invitrogen, Q32851). The sequencing libraries have been subsequently sequenced on an Illumina NextSeq 500 sequencer in paired-end mode.
Sequencing outcomes have been demultiplexed utilizing Illumina bcl2fastq (v.2.20), requiring an ideal match to indexing BC sequences. Low-quality reads or bases and Illumina adapter sequences have been trimmed utilizing Trimmomatic (v.0.39). Reads have been then mapped to the mouse genome (mm10) utilizing bwa (v.0.7.17). The protection and mutations in and across the Tbxt locus have been checked via visualization in a mirror model of the UCSC Genome Browser.
Mouse experiments and producing Tbxt
Δexon6/+ mice
All mouse experiments have been carried out following NYULH’s animal protocol tips and carried out on the NYU Langone Well being Rodent Genetic Engineering Laboratory. Mice have been housed within the NYU Langone Well being BSL1 barrier facility in a 12-h mild to 12-h darkish cycle, with ambient temperature and humidity circumstances. All experimental procedures have been authorized by the Institutional Animal Care and Use Committee at NYU Langone Well being. Wild-type C57BL/6J (pressure 000664) mice have been obtained from The Jackson Laboratory.
The TbxtΔexon6/+ heterozygous mouse mannequin was generated via zygotic microinjection of CRISPR reagents into wild-type C57BL/6J zygotes (Jackson Laboratory pressure 000664), adapting a beforehand printed protocol57. Briefly, Cas9 mRNA (MilliporeSigma, CAS9MRNA), artificial information RNAs and single-stranded DNA oligonucleotide have been co-injected into 1-cell stage zygotes following the described procedures57. Artificial information RNAs have been ordered from Synthego as their customized CRISPRevolution sgRNA EZ equipment, with the identical concentrating on websites as used within the CRISPR deletion experiment of mouse ES cells (AUUUCGGUUCUGCAGACCGG and CAAGAUGCUGGUUGAACCAG). The co-injected single-stranded DNA oligonucleotide is identical as described above. Injected embryos have been then in vitro cultured to the blastomeric stage, adopted by embryo transferring to the pseudopregnant foster moms. Following zygotic microinjection and transferring, founder pups have been screened based mostly on their irregular tail phenotypes. DNA samples have been collected via ear punches at about day 21 for PCR genotyping and Seize-seq validation to exclude off-targeting on the Tbxt locus.
After confirming the genotype, TbxtΔexon6/+ founder mice have been backcrossed with wild-type C57B/6J mice for producing heterozygous F1 mice. Owing to the numerous tail phenotypes, intercrossing between F1 heterozygotes have been carried out in two classes: sort 1 intercrossing included at the very least one father or mother having no tail or a brief tail, whereas sort 2 intercrossing have been mated between two long-tailed F1 heterozygotes (Desk 1). As summarized in Desk 1, each varieties of intercrossing produced heterogeneous tail phenotypes in F2 TbxtΔexon6/+ mice, thereby confirming the unfinished penetrance of tail phenotypes and the absence of homozygotes (TbxtΔexon6/Δexon6). Grownup mice (>12 weeks) have been anaesthetized for X-ray imaging of vertebra utilizing a Bruker In-Vivo Xtreme IVIS imaging system. To substantiate the embryonic phenotypes in homozygotes, embryos have been dissected at E11.5 gestation stage from the timed pregnant mice utilizing a normal protocol.
Producing Tbxt
insASAY and Tbxt
insRCS2 mice
The engineered TbxtinsASAY and TbxtinsRCS mouse ES cells have been injected into both C57BL/6J-albino (Charles River Laboratories, pressure 493) blastocysts for chimeric F0 founder mice or injected into B6D2F1/J (a F1 hybrid between C57BL/6J feminine and DBA/2J male, Jackson Laboratory pressure 100006) tetraploid blastocysts for homozygote F0 founder mice manufacturing. The tetraploid complementation technique aimed to generate homozygous mice with the proposed genotype within the F0 technology58. By a number of trials of injection utilizing each mouse ES cell traces, we achieved just one TbxtinsASAY/insASAY F0 founder mouse (male) however none for the TbxtinsRCS mouse. Nonetheless, throughout genotype screening for TbxtΔexon6/+ founder mice, we serendipitously recognized a male gray mouse that integrated a heterozygous insertion in intron 5. Genotype evaluation revealed that the insertion was a 220 bp DNA sequence from intron 6 of Tbxt (chromosome 17: 8439335–8439554, mm10), inserted in a reverse complementary state of affairs into intron 5 at a designed CRISPR concentrating on website (chromosome 17: 8438386, mm10). The inserted sequence insRCS2 in intron 5 subsequently varieties a 220 bp inverted complementary sequence pair with its unique sequence in intron 6 (chromosome 17: 8439335–8439554, mm10), resembling the designed TbxtinsRCS and TbxtinsASAY gene constructions. This genotype was subsequently referred to as TbxtinsRCS2. Seize-seq genotyping of this TbxtinsRCS2/+ mouse confirmed that the TbxtinsRCS2 allele is within the C57BL/6 background, whereas the wild-type Tbxt locus of the TbxtinsRCS2/+ founder mouse is from the DBA/2J background. This TbxtinsRCS2/+ mouse was subsequently backcrossed to C57BL/6 wild-type mice and additional intercrossed between F1 heterozygotes to provide homozygotes (TbxtinsRCS2/insRCS2) within the F2 technology. Seize-seq evaluation of TbxtinsRCS2/insRCS2 mice confirmed their C57BL/6 background on the Tbxt locus (Prolonged Information Fig. 8). We additionally in contrast the tail phenotypes in age-matched C57BL/6 and DBA/2J mice and located no distinction (information not proven), which instructed that any genetic background distinction between the 2 strains doesn’t have an effect on tail size. The TbxtinsRCS2 mice (each heterozygotes and homozygotes) have been subsequently used for the evaluation of tail phenotypes.
The TbxtinsASAY and TbxtinsRCS2 founder mice, each male, have been individually backcrossed to wild-type C57B/6J mice for producing heterozygous F1 pups, adopted by intercrossing between F1 heterozygotes to generate homozygotes in F2 technology. With all genotypes out there, mouse tail lengths have been measured month-to-month throughout genotypes and intercourse teams. Moreover, two varieties of breading pairs, TbxtinsRCS2/+ × TbxtΔexon6/+ and TbxtinsRCS2/insRCS2 × TbxtΔexon6/+, have been carried out throughout completely different founder traces of TbxtΔexon6/+ mice to analyse tail phenotypes of their offspring. These outcomes are summarized and introduced in Desk 2.
To analyse the isoform expression patterns of mouse Tbxt within the embryonic tailbud area, wild-type, heterozygote and homozygote embryos from intercrossing experiments (TbxtinsRCS2/+ × TbxtinsRCS2/+, TbxtinsASAY/+ × TbxtinsASAY/+) have been dissected on the E10.5 gestation stage. The tailbud for every embryo was collected for isolating complete RNA, and along with embryonic tissue for gDNA for use for genotyping. These outcomes are introduced in Fig. 4e.
Splicing isoform detection
Whole RNA was collected from undifferentiated and differentiated cells of each human and mouse ES cells, and from embryonic tailbud samples, utilizing a normal column-based purification equipment (Qiagen RNeasy Package, 74004). DNase therapy was utilized throughout RNA extraction to take away any potential DNA contamination. Following extraction, RNA high quality was assessed via electrophoresis based mostly on ribosomal RNA integrity. Reverse transcription was carried out utilizing 1 µg of high-quality complete RNA for every pattern and a Excessive-Capability RNA-to-cDNA equipment (Utilized Biosystems, 4387406). DNA oligonucleotides used for RT–PCR and/or quantitative RT–PCR are listed in Supplementary Desk 1.
Transcriptomics analyses in differentiated mouse ES cells
Whole RNA samples remoted from day-1 in vitro-differentiated mouse ES cell traces throughout wild-type, TbxtinsASAY/insASAY, TbxtΔexon6/+ and TbxtΔexon6/Δexon6 genotypes have been used for bulk RNA sequencing evaluation. RNA samples have been ready utilizing a normal column-based purification equipment (Qiagen RNeasy equipment, 74004). Two organic replicates have been ready for every mouse ES cell genotype, with the 2 TbxtΔexon6/Δexon6 mouse ES cell samples coming from completely different clones. RNA sequencing libraries have been ready utilizing a NEBNext Extremely II Directional RNA Library Prep equipment (NEB, E7765L) via its polyA mRNA sequencing workflow by utilizing the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB, E7490L).
Uncooked sequencing reads have been mapped to the mouse genome (mm10) with STAR (v.2.7.2a) aligner59. The resultant strand-specific learn counts of all samples have been built-in right into a matrix for downstream evaluation. Differentially expressed genes have been detected utilizing DESeq2 (v.1.40.2)60, utilizing its default two-sided Wald take a look at with the cut-off of log2(fold expression change) > 0.5 and a number of test-adjusted P worth < 0.05. The highest 500 variable genes from DESeq2 throughout all samples have been used to carry out principal part evaluation. The Tbxt goal genes have been obtained from a earlier publication41, outlined by important Tbxt ChIP–seq peak alerts detected in in vitro-differentiated mouse ES cells. The set of Tbxt goal genes was intersected with the numerous differentially expressed genes recognized in every mutant pattern in contrast with the wild-type controls, and these have been aggregated to generate the general set of differentially expressed Tbxt goal genes throughout the analysed mouse ES cell traces. These differentially expressed Tbxt goal genes have been visualized utilizing a heatmap, with the log10-transformed normalized transcript matrix adopted by z rating standardization throughout samples.
Reporting abstract
Additional info on analysis design is offered within the Nature Portfolio Reporting Summary linked to this text.